


- Startpagina tijdschrift
- Volume 14 (2010)
- numéro 1
- Optimized electroporation-induced transformation in Microcystis aeruginosa PCC7806


Weergave(s): 2757 (1 ULiège)
Download(s): 264 (1 ULiège)

Optimized electroporation-induced transformation in Microcystis aeruginosa PCC7806

Nota's van de redactie
Received on March 27, 2009; accepted on September 11, 2009
Résumé
Transformation par électroporation réussie chez Microcystis aeruginosa PCC7806. La disruption génique chez les cyanobactéries est difficile et constitue un obstacle pour les manipulations génétiques. Les quelques rares articles qui font état de ce problème mentionnent des méthodes généralement peu précises et difficilement reproductibles. Nous décrivons ici une transformation par électroporation réussie chez Microcystis aeruginosa PCC7806 pour lesquelles les conditions de réussite pour l'électroporation et la transformation sont analysées.
Abstract
Gene disruption in cyanobacteria is difficult and comprises an obstacle for genetic manipulation. Very few reports tackled this problem but the methods used are usually obscure and hardly reproducible. Here we describe an optimized electroporation-induced transformation in Microcystis aeruginosa PCC7806 where conditions for successful electroporation and transformation are investigated.
1Gene disruption is a technique of genetic manipulation where insertion of antibiotic gene cassette within coding region of a certain gene causes loss of that gene’s function through homologous recombination (Porter, 1986). However, this is of rare occurrence in cyanobacteria due to the activity of endo- and exonucleases (Porter, 1986). Therefore, the establishment of an easy-to-follow gene disruption protocol in cyanobacteria to study genes’ functions is necessary. Microcystins are hepatotoxins produced through the activities of two genetic systems: NRPS (non-ribosomal peptide synthetases) and PKS (polyketide synthases) (Tillett et al., 2000). The gene studied here is mcyB, one of the peptide synthetase genes essential for microcystin biosynthesis. Insertional mutagenesis of this gene resulted in no detectable toxin production (Dittmann et al., 1997). However, the latter protocol persistently failed even though it was used on the same isolate i.e. Microcystis aeruginosa PCC7806. Hence the procedures were scrutinized and an alternative protocol was developed. The present protocol requires no complicated techniques and can possibly be applied on different genes.
2In order to verify the essence of mcyB gene for toxin biosynthesis, a gene disruption experiment was performed. Primers designed by Dittmann et al. (1997): QF (TTGAGCAAGGACAATTGC) and HR (CTCCCGCATAATCACAACAG) were initially used to amplify the mapep1 fragment for the purpose of knock-out of mcyB in M. aeruginosa PCC7806. Despite the numerous trials, amplification persistently failed. Therefore, other primers were designed to amplify a different region within mcyB (position 18256-20891) to allow future manipulation. The primer sequences were Mcyfor2 (CACCCCCCTGAGGGTGGACAGACTCC) and Mcyrev2 (GAGGGTGGAAACAATATGATAAGCTA). This would amplify a 2636 bp fragment within mcyB. A restriction analysis of the fragment was conducted using DNAman package, Lynnon,1999.The PCR mixture contained the following components: 1 µl MgCl2, 0.5 µl DMSO, dNTP at a final concentration of 200 µM each, primer forward and reverse at 200 µM each, 1 × Phusion buffer and 1U of Phusion polymerase enzyme (Finnzymes, Finland.) Genomic DNA was extracted using the DNeasy extraction kit (Qiagen, The Netherlands). Five µl containing 100 ng genomic DNA was used as a template in a total volume 50 µl reaction mixture. Touch-down proved to be the most efficient protocol than any other protocol tested. The thermal programme was as follows: Initial: 95°C, 5 min x 1 cycle; Main: 94°C, 1 min, 64°C, 1.5 min, 72°C, 1.5 min x 2 cycles; 94°C, 1 min, 62°C, 1.5 min, 72°C, 1.5 min x 2 cycles; 94°C, 1 min, 60°C, 1.5 min, 72°C, 1.5 min x 2 cycles; 94°C, 1 min, 58°C, 1.5 min, 72°C, 1.5 min x 2 cycles; 94°C, 1 min, 56°C, 1.5 min, 72°C, 1.5 min x 30 cycles and Final: 72°C, 10 min x 1 cycle. The PCR product was first ligated to pENTR-D-TOPO vector (Invitrogen, The Netherlands). The transformation into competent Escherichia coli (Top 10) cells was performed according to the manufacturer’s instructions. Plasmid-containing insert was purified using Qiagen plasmid kit and digested using HindIII restriction enzyme. The restriction digest contained 0.8 µl HindIII restriction enzyme, 1 µl 10 × buffer 2 (New England Biolab, UK), 0.2 µl bovine serum albumin, 2 µl of plasmid in a total volume 10 µl and was incubated at 37°C for 1 h. The reaction was terminated by incubation at 65°C for 10 min. The antibiotic gene cassette used was chloramphenicol fragment, about 750 bp, which was kindly supplied by Dr Colin Lazarus, University of Bristol, UK. The fragment had HindIII restriction sites inserted by PCR at both ends. The 5 µl of the digest were directly ligated to 6 µl of the chloramphenicol fragment, using 2 µl of T4 ligase and its buffer at 1 x concentration in 20 µl ligation reaction. The ligation mixture was incubated at room temperature overnight then used to transform E. coli competent cells (Top 10, Invitrogen). Selection for transformants was performed on chloramphenicol (30 µg.ml-1)–supplemented agar plates. The construct was purified and its concentration was determined spectrophotometrically at λ 260 nm. Twenty µl of the construct were then methylated in the presence of 1 × S-adenosyl methionine, 1 × NEB buffer number 2, one U of the enzyme CpG methylase (New England Biolabs) in 100 µl total volume. Half the amount of construct was also used in a separate experiment to test for the optimum plasmid DNA concentration (Dittmann et al., 1997). The mixture was incubated for 3 h at 37°C. Ten ml sample of cultures both at logarithmic and late stationary phase were centrifuged and used in different transformation techniques as follows:
3– natural transformation as described by Dittmannet al. (1997);
4– electroporation-induced transformation.
5Cells of wild type M. aeruginosa PCC7806 at both mid logarithmic and stationary stages were centrifuged and concentrated at a density of 10-9 cells per ml.
6The cell pellet was washed three times in 500 µl of 0.1 mM cold HEPES (N-2-hydroxyethylpiprazine-N'-2-ethanesulfonic acid, pH 7.2) then chilled on ice. The methylated construct (100 µl) was added to M. aeruginosa cells on ice to allow for a pre-pulse contact at a low temperature for 1-2 h. Different pre-electroporation treatments were also tried. For example, Microcystis cells were washed several times in serial volumes of 10% ice cold sterile glycerol instead of HEPES. Additionally a higher concentration of HEPES buffer 1 mM was also tested. The cells were electroporated using BioRad Gene Pulser equipped with a 25-µF capacitator and resistance was fixed at 400 ohms. Forty µl of cell-construct suspension were electroporated in a pre-chilled, sterile cuvette with a 2-mm gap between the electrodes. Different field strengths were used (Figure 1). Field strength is calculated as voltage applied divided by gap distance of the cuvette (0.2 cm) (Thiel et al., 1989). Immediately after single electric pulse, 2 ml of cold sterile BG 11 medium was added to the 40 µl electroporated mixture and mixed well. The diluted electroporated cells were first cultured in 10 ml BG 11 without chloramphenicol, at 40 µmol.m-2.s-1 continuous white light for one day. The cultures were then centrifuged and the pellet was directly streaked out on solidified BG11 supplemented with chloramphenicol. The cultures were grown at room temperature at 14 µmol.m-2.s-1 continuous white light and the plates were checked daily under the dissecting microscope (Leica, Germany). After one month, the number of transformants were counted and transformation efficiency was calculated using the following equation: Transformation efficiency (%) = the number of transformants / the total number of cells before transformation x 100.
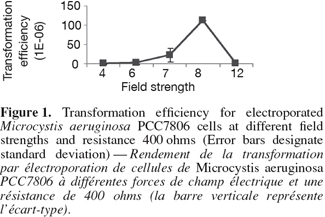
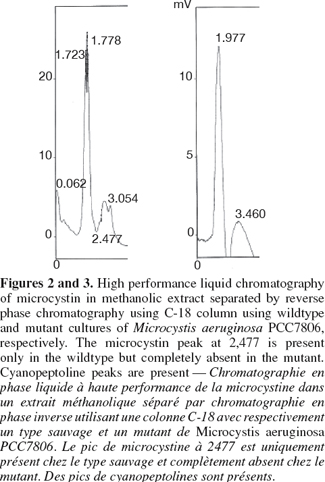
7The colonies were picked up after one month under the dissecting microscope, placed in BG11 supplemented with chloramphenicol and grew under same growth conditions. The previous steps were repeated for three months to allow complete homozygosity of mutants. Mutants were tested for the integration of the disrupted gene using forward primer of chloramphenicol fragment TTGGCGGCCGCATTAGGC and reverse primer of the mcyB homologous fragment Mcyrev2: GAGGGTGGAAACAATATGATAAGCTA. The same PCR composition and touch-down protocol were followed. For Reverse phase HPLC C18 column, microcystin was extracted from lypholised Microcystis cells from both wild type and mutants using 80% methanol in dark bottles for two days. The suspension was centrifuged at 14,000 x rpm and the supernatant was used for reverse phase HPLC (Perkin-Elmer Phenomenex HPLC, C18 Ultracarb 7.0 Column ODS 20). The mobile phase consisted of two solvent systems:
8– solvent A: water with 0.05% trifluoracetic acid;
9– solvent B: acetonitrile with 0.05% trifluoracetic acid.
10The elution programme proceeded as follows: 70% solvent A; 30% solvent B for 5 min; and then decreasing the concentration of solvent A by 5% while increasing solvent B by 5% every 5 min until reaching concentration 50% of solvent A: 50% solvent B. Total run time was 30 min, flow rate 1 µl.min-1 and UV detector set at 238 nm.
11The experimental procedures began with amplifying the mapep1 fragment from isolate M. aeruginosa PCC7806. However, the numerous amplification trials using the QF and HR primers (Dittmann et al., 1997) persistently failed. The sequence of those primers was checked against sequence of M. aeruginosa PCC7806 deposited at the GenBank database (accession number AF183408). The BLAST search revealed that the full QF sequence only showed 94% identity to mcyA position (44855-44872) sequence of isolate M. aeruginosa PCC7806 (accession number AF183408) with only a single mismatch. The other primer, HR was only partially homologous to mcyC sequence in the position (54284-54268). None of the primers matched perfectly or fully the sequence of mcyB in M. aeruginosa PCC7806 and the mismatches were mostly at the growing 3'-end. Additionally, the priming sites of the QF and HR primers are nearly 20.57 Kbp apart which makes it impossible to amplify only a 2.8 Kbp fragment as was reported by Dittmann et al. (1997). Therefore, the failure of all amplification trials is due to lack of priming site of those primers in the mcyB. Instead we designed new primer pair to target a different fragment within mcyB of M. aeruginosa PCC7806. Four important considerations were taken into account when designing the primers, choosing the recombinant fragment and forming the construct. First, the region upon which the forward primer is based, must start with a four base pair sequence (CACC) which marks the 5'-end of the forward primer. This is important for cloning the amplicon into plasmid vector pENTER-D-TOPO which uses this specific sequence for directional cloning within vector. Second, the target area must span an open reading frame of no less than 2,000 bp if not more in order to increase chances of success of homologous recombination. Third, the target area must contain a single restriction site for a restriction enzyme that is not found in the cloning vector used in the construct. This is to allow single cut within the insert only. Fourth, the antibiotic gene cassette must be treated with the same restriction enzyme, hence HindIII sites were inserted at both ends of the antibiotic fragment, to generate sticky ends compatible with those generated through restriction of the insert. Accordingly, the target area was specifically chosen so a single HindIII restriction site would be present nearly midway of the DNA fragment, hence allowing considerable long flanking regions on either sides of the restriction site where the antibiotic gene would be inserted. Zang et al. (2007) reported the complete failure of transformation of Synechocystis sp. PCC6803 when a single homology region was found on only one side of the inserted antibiotic gene. The restriction mapping revealed the absence of AvaI endonuclease restriction site in the amplified fragment. This enzyme is found in M. aeruginosa PCC7806 and is responsible for degrading foreign DNA. The same enzyme represents a restriction barrier in Anabaena (Thiel et al., 1989). Choosing the fragment that lacks this restriction enzyme site helped in facilitating construct uptake by the host. The restriction mapping also revealed the presence of a single restriction site for HindIII restriction enzyme only 1,600 bp far from the beginning of the target fragment. The restriction digestion with HindIII of the plasmid carrying the insert resulted in the linearization of the construct into a single fragment to which the chloramphenicol resistance gene was ligated. The touch-down protocol allowed the high specificity of primer binding and eliminated the non-specific products due to the application of high annealing temperatures. The PCR amplicon needed to be blunt-ended to allow cloning into the plasmid cloning vector pENTR-D-TOPO. Therefore the Phusion enzyme which generates blunt ended amplicons, was used. The use of cloning vector pENTR-D-TOPO has the advantage of absence of commercial polylinker site that is found in most widely-used vectors. Hence it lacks, HindIII restriction site which occurs solely in the insert, thus allowing single restriction and ligation event within the digested insert to place the antibiotic gene. Also the lack of polylinker site, commonly present in most commercial vectors, served to reduce the action of host nucleases that recognize restriction sites on the foreign DNA i.e. construct, and degrade it. Nevertheless, there are a few restriction sites on this vector that are of relatively rare occurrence such as NotI and AscI, NruI, HpaI and PvuII and were not reported to occur in M. aeruginosa PCC7806 (Frangeul et al., 2008). The construct was initially hosted by top 10 E. coli competent cells which are already dam- and dcm-type strain, thereby allowing in vivo methylation of the construct. Using 1 µg of plasmid DNA amount for each unit of CpG methylase and proceeding by natural transformation (Dittmann et al., 1997) was eventually unsuccessful. However, the methylated construct was only successful when double the amount of plasmid DNA was used in subsequent electroporation-induced transformation. For electroporation, the several washing steps with low concentration of HEPES buffer may have helped in removing some of the exonucleases of the host and minimizing the amount of salts present with the construct which is consequently helpful for the electroporation step. The increase in HEPES concentration resulted in failure of transformation as similarly found by Kim et al. (1996). Only logarithmic cultures were amenable to electroporation which is possibly due to the more amenability of cell membranes of young new cells at the logarithmic phase to electroporation than those at the stationary phase (Kim et al., 1996). Pre-pulse incubation at low temperature allowed contact between foreign (construct) DNA and host cells and increased chances of construct uptake. The electroporation was only successful for cultures at logarithmic phase using high resistance, i.e. 400 ohms, and not at low resistance, i.e. 100 ohms. At 400 ohms, the field strengths arranged according to their frequency of transformation were 8 followed by 7, then 6 kV.cm-1. Field strength 12 did not result in any transformation (Figure 1). Therefore, intermediate field strengths at high resistance would be optimum for transformation possibly due to their high killing rate and to the size of cyanobacteria (Thiel et al., 1989). Testing the mutants for the integration of the disrupted gene within the chromosome using PCR resulted in a product of approximately 1,700 bp. The comparison of the HPLC profiles for the methanolic extract of wild type M. aeruginosa PCC7806 to that of the mutant confirmed the lack of any detectable microcystin in the latter (Figures 2 and 3) as was further confirmed by HPLC reference library. Taken all together, the success of electroporation-induced transformation and consequently gene disruption relies mainly on: the choice of suitable long homologous fragment and vector with limited restriction sites; the methylation of a high concentration of the construct, the pre-pulse treatments of logarithmic phase cultures and construct at low temperature, the electroporation at intermediate field strengths and high resistance level and the appropriate incubation conditions.
12Acknowledgements
13The author is thankful to Professor Paul Hayes, Dr Colin Lazarus and Dr Maged Mohamed, School of Biological Sciences, University of Bristol (UK) for their suggestions. The author also acknowledges gratefully the receipt of UNESCO priority programme II fellowship and COMSTECH-ISESCO grant.
Bibliographie
Dittmann E. et al., 1997. Insertional mutagenesis of a peptide synthetase gene that is responsible for hepatotoxin production in the cyanobacterium Microcystis aeruginosa PCC7806. Mol. Microbiol., 26(4), 779-787.
Frangeul L. et al., 2008. Highly plastic genome of Microcystis aeruginosa PCC7806, a ubiquitous toxic freshwater cyanobacterium. BMC Genomics, 9, 274-294.
Kim W.-J., Heo T.-R. & So J.-S., 1996. Optimised transformation by electroporation of Lactococcus lactis IL 1403. Biotechnol. Techn., 10(8), 553-558.
Porter R.D., 1986. Transformation in cyanobacteria. Crit. Rev. Microbiol., 13, 111-132.
Thiel T., 1994. Genetic analysis of cyanobacteria. In: Bryant D.A., ed. The molecular biology of cyanobacteria. Dordrecht, The Netherlands: Kluwer Academic Publishers, 581-611.
Thiel T. & Poo H., 1989. Transformation of a filamentous cyanobacterium by electroporation. J. Bacteriol., 171, 5743-5746.
Tillett D. et al., 2000. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC7806: an integrated peptide-polyketide synthetase system. Chem. Biol., 7, 753-764.
Zang X. et al., 2007. Optimum conditions for transformation of Synechocystis sp. PCC6803. J. Microbiol., 45(3), 241-245.
Om dit artikel te citeren:
Over : Nermin Adel El Semary
Helwan University. Faculty of Science. Department of Botany and Microbiology. Ain Helwan campus. ET-11795 Helwan (Egypt). E-mail: nerminel_semary@helwan.edu.eg
