


- Startpagina tijdschrift
- Volume 20 (2016)
- numéro 4
- The hunt for original microbial enzymes: an initiatory review on the construction and functional screening of (meta)genomic libraries


Weergave(s): 3934 (30 ULiège)
Download(s): 454 (13 ULiège)

The hunt for original microbial enzymes: an initiatory review on the construction and functional screening of (meta)genomic libraries

Nota's van de redactie
Received on February 24, 2016; accepted on June 16, 2016
Résumé
La chasse aux enzymes microbiennes originales: une synthèse bibliographique initiatique sur la construction et le criblage fonctionnel de banques (méta)génomiques
Introduction. La découverte de nouvelles enzymes est tout aussi primordiale pour les sciences appliquées que pour les sciences fondamentales. Dans ce contexte, les enzymes microbiennes, connues pour être extrêmement diversifiées et faciles à produire, sont fort recherchées par diverses approches.
Littérature. Cette synthèse bibliographique débute avec une comparaison des approches qui utilisent la mise en culture pour rechercher des enzymes microbiennes avec celles indépendantes de la mise en culture de microorganismes. Au sein de ces deux types d’approches, nous discutons également les avantages et désavantages des méthodes qui se basent sur les séquences et de celles qui se basent sur l’observation d’une activité. Nous discutons ensuite les différents facteurs qui peuvent influencer la réussite de la construction et du criblage d’une banque génomique ou métagénomique : l’environnement étudié, l’extraction et la restriction de l’ADN, le type de vecteur utilisé (plasmide, cosmide, fosmide, BAC ou vecteur navette) et la cellule hôte eucaryote ou procaryote choisie.
Conclusions. La construction et le criblage de banques sont délicats et demandent de l’expertise. Ils peuvent être optimisés en combinant différentes approches, comme travailler avec les microorganismes cultivables et les non cultivables, rechercher des enzymes sur base de la séquence et de leur fonction ou encore faire des criblages dans différentes cellules hôtes. Toutes ces étapes permettent d’élargir la quantité et la diversité des enzymes isolées à partir d’un échantillon environnemental.
Abstract
Introduction. Discovering novel enzymes is of interest in both applied and basic science. Microbial enzymes, which are incredibly diverse and easy to produce, are increasingly sought by diverse approaches.
Literature. This review first distinguishes culture-based from culture-independent methods, detailing within each group the advantages and drawbacks of sequence- and function-based methods. It then discusses the main factors affecting the success of endeavors to identify novel enzymes through construction and functional screening of genomic or metagenomic libraries: the sampled environment, how DNA is extracted and processed, the vector used (plasmid, cosmid, fosmid, BAC, or shuttle vector), the host cell chosen from the available prokaryotic and eukaryotic ones and the main screening steps.
Conclusions. Library construction and screening can be tricky and requires expertise. Combining different strategies, such as working with cultivable and non-cultivable organisms, using sequence- and function-based approaches, or performing multihost screenings, is probably the best way to identify novel and diverse enzymes from an environmental sample.
Inhoudstafel
1. Introduction
1In both applied and basic science, there is currently great interest in identifying and producing novel enzymes and biocatalysts. On the one hand, this could contribute to develop green industrial applications and white biotechnologies (Gavrilescu et al., 2005), while on the other hand, the discovery of genes encoding for novel enzymes and of novel functions can help us understand specific ecosystems (Ufarte et al., 2015). Furthermore, the study of original enzymes with novel three-dimensional structures or catalytic mechanisms can shed light on the complex relationships between protein structure and function (Ufarte et al., 2015).
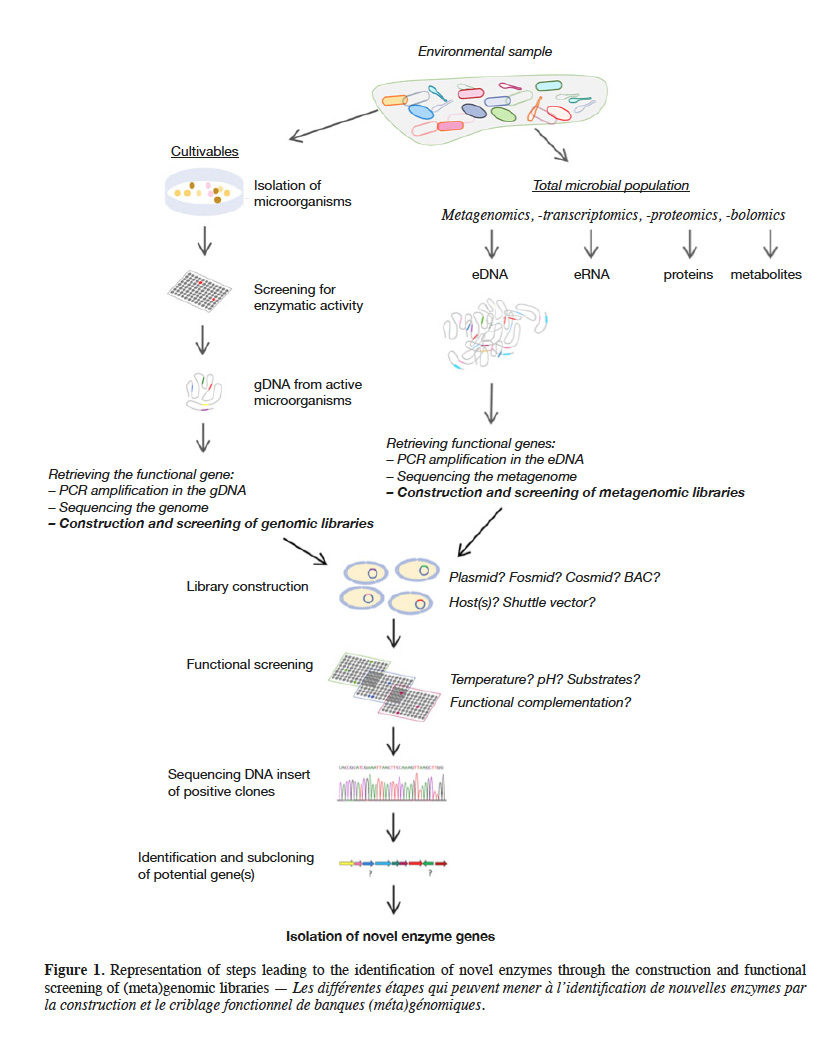
2Microorganisms are the greatest and most studied source of enzymes, mainly because they are easy to manipulate and to produce in large scales. In addition, their enzymes are biochemically diverse and have broad range of activities facing variation in environmental parameters as pH, temperature and salinity (Adrio et al., 2014). To discover novel microbial enzymes, diverse types of functional analysis can be applied either to microorganisms themselves or to microbial genomes. In this review, we highlight the different ways in which DNA library screening can lead to identify novel genes, enzymes, protein families, and functions. We first briefly place the different techniques used for this purpose in their respective contexts, distinguishing culture-based from culture-independent methods. We then discuss the factors liable to limit the output of these approaches: the sampled environment, the chosen vector and DNA insert size range, the paucity of available host cells, and certain crucial or optional steps performed during functional screening. The chart on figure 1 provides an outline of these methods. For complementary information on the various topics broached, readers can refer to the most recent reviews cited throughout this publication.
2. Traditional and current techniques
3Towards the end of the 19th century, researchers discovered that certain natural proteins, for which the term “enzyme” was coined, act as biocatalysts. They also became aware of the potential use of enzymatic catalysis to replace chemical catalysis, and set out to develop such applications, using either whole cultivable cells or (partially) purified preparations of natural enzymes. Efforts then focused on finding or creating enzymes with improved features. In the 1990s, directed evolution emerged as a novel means of improving known enzymes. It involves generating from a microbe producing a protein of interest a library of mutants by random approaches and then screen the library for specific and better activity, selectivity, and/or stability (Cobb et al., 2013). It now includes a package of traditional and modern mutation strategies for improving or altering the activity of known biocatalysts (for recent reviews, see Denard et al., 2015; Packer et al., 2015). Another milestone was the advent of metagenomics, the culture-independent genomics of entire microbial consortia present in environmental samples. Metagenomics was first used to assess bacterial diversity through phylogenetic analysis of 16S rRNA sequences and to answer the question “who is in there?”. It rapidly gained a more functional dimension, with attempts to answer more difficult questions: “what are they doing?” or “what can they do?” (Handelsman, 2004).
4Microorganisms in an environmental sample include a small minority of cultivable ones and a huge majority of not-yet-cultivable microorganisms. Approaches to identify novel enzymes from each of these groups are described below.
2.1. Culture-dependent or independent approaches
5Enzymes have long been recovered from cultivable microorganisms exhibiting specific activities. Microorganisms isolated from an environmental sample are screened in liquid or solid medium for activities of interest. Active natural isolates can then be used directly in bioreactors, either to produce the enzyme or to catalyze an industrial reaction (Roberts et al., 1995). However, optimizing the process can require countless adjustments, diverse side reactions might dominate or interfere with the substrate, and a product or co-solvent might disrupt the enzyme (Roberts et al., 1995). An alternative approach is to clone the enzyme-encoding gene into a well-known host cell whose behavior can be controlled. To retrieve the gene of interest sequence-based and function-based approaches can be used (discussed below). In contrast, culture-independent approaches (working with non-cultivable organisms or total microflorae) always involve, as a first step, extraction of nucleic acids or gene products. According to what is extracted from an environmental sample, i.e. total microbial DNA, RNA, proteins, or metabolites, researchers speak of metagenomics, metatranscriptomics (Warnecke et al., 2009), metaproteomics (Schofield et al., 2013), or metabolomics (Prosser et al., 2014). At the time of sampling, the last three disciplines mentioned retrieve only gene transcripts or produced proteins or metabolites. They are mainly used to understand functional interactions and discover novel metabolic pathways. Here we will focus solely on metagenomics. This discipline also includes sequence-based and function-based approaches (discussed below).
2.2. Sequence-based approaches
6To retrieve a gene encoding for an enzyme of interest in genomic DNA (gDNA) or environmental DNA (eDNA), one can either amplify it by Polymerase Chain Reaction (PCR), using primers designed from sequence motifs found in similar enzymes, or identify it by sequencing the entire microbial gDNA or eDNA (shot-gun or DNA library sequencing) and comparing its sequences against genomic databases. This last method was unthinkable before, as Sanger sequencing was costly and very time consuming. Fortunately, sequencing has become less expensive in recent years, and results are now rapidly obtained thanks to second (e.g. 454, MiSeq Illumina, Ion torrent) and third (e.g. PacBio) generation sequencing methods (reviewed in Bleidorn, 2015; Faure et al., 2015; Rhoads et al., 2015). Yet even though processing of sequence data has been simplified more and more by progress in bioinformatics, it can be difficult or time consuming to choose a specific enzyme in the immensity of generated data, to predict the characteristics of identified putative enzymes or whether a protein will be produced easily in cultivable host cells for further analysis. These sequence-based approaches are possible only if the enzymes sought are closely related to known ones; they cannot lead to the discovery of completely novel enzymes or enzyme families. Finally, this type of approach can also yield false hits, due to the numerous wrong annotations found in non-curated databases. Sequence-based methods are therefore used mostly to explore the microbial diversity of an environment on the basis of 16S or 18S rRNA gene sequences or to understand the gene arrangement in a microbial genome.
2.3. Function-based approaches
7The gDNA from a microorganism of interest or the eDNA from a studied environment might be used to construct (meta)genomic libraries in a well-known cultivable host cell, and then screening these libraries for clones displaying the sought enzymatic activity. Functional (meta)genomics, which relies solely on gene function rather than sequence similarities, has a considerable advantage when applied to novel bacterial taxa (strains, species or genera) or unknown bacteria, since it has a high probability of yielding genes encoding novel enzymes. In addition, it is possible to screen for specific enzymatic characteristics by varying the screening conditions (e.g. temperature, pH and substrate concentration). Lastly, if an enzyme-encoding gene is recovered by activity screening, the protein should be readily produced in the well-known host used for library construction. Functional (meta)genomics has already led to the discovery of extraordinary novel biocatalysts from all around the world and to assigning numerous “hypothetical proteins” in databases (Ferrer et al., 2016). Nevertheless, the screening is very fastidious, particularly when applied to metagenomes (many clones have to be screened to cover a majority of the genes present in an environmental sample). The screening yields are generally low, given the multiple constraints (such as heterologous expression in the chosen host cell, substrate affinity or a missing co-factor) (Ekkers et al., 2012), even more with functional metagenomics because no selection of “active” microorganisms is done upstream from library construction and screening (which can be realized while working with cultivable microorganisms). Therefore functional metagenomics is recommended for work on low-density populations of microbes that are hard to grow. Robotized high-throughput screening may also considerably enhance the number of screened clones and inevitably the number of positive hits. Function-based approaches on cultivable microorganisms can yield enzymes closely related to those of other cultivable organisms. To maximize the yield and the novelty of the resulting discoveries, it is therefore advisable to exploit underexplored environments and/or to develop novel growth media, conditions and/or techniques of isolation (Highlander, 2014; Kamagata, 2015). Over the past decade, innovations have emerged in the culture and isolation of microorganisms (reviewed in Pham et al., 2012). Cycling cultures implies cyclical varying culture and growth conditions (Dorofeev et al., 2014). Culture in microwells with phenotypic microarrays are used to screen for and identify optimal growth conditions (Borglin et al., 2012). In situ techniques are also developed to enhance interactions with the environment and the other microorganisms living in it (Jung et al., 2014; Steinert et al., 2014). The use of novel isolation media will lead to the identification of unknown bacterial taxa and hence to the discovery of exciting novel enzymes.
3. Sampled environments
8Microorganisms are found in every single environment on earth and must obviously produce enzymes enabling them to survive wherever they live (Yarza et al., 2014). Therefore, the functional analysis of each microbial niche should contribute to the knowledge on how ecosystems work and lead to identifying original functional genes and enzymes. The choice of an environment to be prospected will depend on the type of enzyme one seeks and on the desired features of the identified biocatalysts. All environments are not equal in the manner they should be explored and in the diversity and novelty of the findings they will yield.
9Soils and oceans have been intensely investigated for their microbial diversity. However marine waters have been much less studied by functional analysis. Nevertheless, the microbial diversity of soils and oceans is so immense that these resources still remain undersampled, and their potential as sources of new enzyme discoveries seems infinite. Microbial hotspots and/or hot moments, described respectively by ecologists as spots and short periods of time showing disproportionately high reaction rates relative to the surrounding matrix or to adjacent longer time periods (De Monte et al., 2013; Kuzyakov et al., 2015), could help in choosing the particular habit to be explored in these vast environments and the moment of the sampling.
10Exploring extreme environments has also led to identifying original biocatalysts with unusual characteristics: so-called microbial extremozymes (Raddadi et al., 2015). Recent reviews focus on how to improve screening conditions and yields in the case of samples from cold (Vester et al., 2015) or saline environments (de Lourdes Moreno et al., 2013; Raval et al., 2013), using culture-dependent and -independent methods. Bioprospection for enzymes of other extremophilic microbes, such as piezophiles from deep-sea sediments (Kato, 2012) or (halo)alkaliphiles (Borkar, 2015), is still in its infancy, because of the very specific culturing and screening conditions it requires. Nevertheless, such microorganisms should have huge biotechnological potential.
11In the last decade, functional studies have also focused on gut microbiota, biofilms, and symbionts. Reviews on the subject include for example one devoted to microbes inhabiting the human gut (Walker et al., 2014), one on insect symbionts (Berasategui et al., 2015), one on rumen microbes (Morgavi et al., 2013), and one on algal biofilms (Martin et al., 2014). Interactions between microorganisms and their host are generally intense, and sites where symbiosis occurs are rich in enzymes. Microorganisms living in tight, specialized symbiosis with a host or with other microbes tend not to grow well in culture and should therefore be best suited for functional metagenomics (Handelsman, 2004).
12Finally, naturally or artificially enriched environments (Kamagata, 2015), such as copper-enriched (Riquelme et al., 1997) and oil-fed soils (Narihiro et al., 2014), can also be explored for novel enzyme types with important ecological or industrial applications.
13The good news is that a practically infinite number of environments remain to be tapped for novel enzymes. Even among the environments that have been studied by metagenomics over the last 20 years, it seems that only 11% have been studied with this goal in mind (Ferrer et al., 2016). Functional analysis of samples taken from as yet unexplored habitats is bound to yield original and exceptional microbial biocatalysts.
4. Dna extraction and processing
14Once the environment is chosen, it is necessary to culture microbial cells, screen them for activity, and extract gDNA or to directly extract eDNA (culture-independent approach). The quality of the extracted DNA might be checked on an agarose gel and its quantity and its purity by spectrophotometry (e.g. with the NanoDropTM spectrophotometers). The extracted gDNA or eDNA should not be degraded and be as pure as possible. If the DNA is degraded, its quantity and the average insert size will be affected, and if contaminants (e.g. humic acids coextracted from soil samples [Zhou et al., 1996], host DNA from alga-associated bacteria [Burke et al., 2009], or residual chemicals from the extraction method) remain, it will be hard to achieve enzymatic DNA restriction and ligation or the libraries will be biased. DNA could be purified and size-selected on agarose gel or by ethanol or PEG/NaCl precipitation (He et al., 2013). If eDNA is recovered, the extraction method yield must be high, to not preferentially retain or eliminate some taxa and, thus, to avoid diversity bias (Thomas et al., 2012).
15When the quality of the extracted DNA has been checked, the DNA is digested with restriction enzymes to the desire insert-size (see below small- and large-insert libraries) and to obtain compatible ends for further cloning. Then, the purified fragments are cloned into cloning vectors by enzymatic DNA ligation for introduction into host cells. The restriction enzyme is chosen mainly according to the type of ligation envisaged (blunt or sticky ends), whether and where the extracted DNA is methylated (some restriction enzymes are sensitive to dam, dcm, or CpG methylation), and the desired DNA insert size. Two types of libraries can be constructed: small- and large-insert libraries (reviewed in Kakirde et al., 2010).
16Small-insert libraries contain DNA fragments smaller than 20 kb inserted into plasmids. These vectors have high copy numbers and strong vector-borne promoters, thus favoring higher enzyme production and better activity detection. Small DNA fragments are easily manipulated, ligated into vectors, and introduced into host cells, but working with plasmids is fastidious, as they cover only small fragments of DNA, the screening to find positive clones requires a large number of clones to be analyzed.
17Large-insert libraries are technically harder to construct but have the advantage of providing more information on the phylogenetic affiliation of the DNA insert and the identified functional genes. Furthermore, large inserts favor the identification of enzymes encoded by genes in large clusters or operons and whose synthesis depends on constitutional promoters upstream from the genes of interest. On the other hand, a larger insert is more likely to have a transcription terminator before the gene of interest, and thus to display early transcription termination (Gabor et al., 2004). To prevent this, adequate vectors and host strains have been developed by genetic engineering (Terrón-González et al., 2013). Cosmids and fosmids can accommodate DNA inserts 25 to 50 kb in size, and bigger ones (up to 300 kb) can be cloned into bacterial artificial chromosomes (BACs). Cosmids are artificially constructed vectors containing the Cos site, which permits packaging of DNA into phage lambda for transfection of Escherichia coli. BACs, designed to introduce large DNA inserts into E. coli, are based on the single-copy F plasmid of this bacterium. The inserted DNA is present in low copy number and is thus more stable (Shizuya et al., 1992; Wanga et al., 2014). Fosmids are cosmid-based vectors containing the replication origin of the E. coli F plasmid as well. They thus combine the stability-favoring properties of BACs with easier manipulation (Rodriguez-Valera, 2014). Kits are now available for easy cloning of DNA into fosmids/BACs and even for increasing the copy number of the insert-bearing vector in E. coli. Examples include the cloning kits CopyRight® v2.0 Fosmid (Lucigen, USA), CopyControl™ BAC, and CopyControl™ Fosmid Library (Epicentre, USA).
18When choosing a cloning vector one should also consider the host cell to be used for library construction and screening. If one intends to use different hosts, it could be best to use a shuttle vector or a broad-host-range vector containing more than one replication origin, suitable for expression in various hosts (Martinez et al., 2004; Aakvik et al., 2009).
5. Host cells for library construction and/or screening
19Heterologous expression is a major challenge in functional screening of (meta)genomic libraries. The transformed host cell must be able to express the foreign DNA and ensure proper folding of the resulting protein(s), and this is not easily achieved. Promoter, terminator, and ribosome binding sites can be added to cloning vectors, and expression can be predicted by bioinformatics (Gabor et al., 2004), but some factors affecting transcription, translation, or the state of a protein in the host cell can be problematic and impossible to control. For example, rare codons unrecognized by the host cell can lead to ineffective translation, production of truncated polypeptides or formation of inclusion bodies after translation, resulting in insoluble and inactive proteins.
5.1. Host-cell characteristics for functional (meta)genomics
20Host cells for constructing DNA libraries are not easy to find, because they must meet many requirements.
21– Being transformable (i.e. having natural competence) is not enough; they should have high transformation yields. When constructing libraries, numerous unique recombinant plasmids must be introduced.
22– Microbial cells do not easily accept and express foreign DNA. The host cells should thus be genetically accessible and modifiable. They generally contain mutations affecting the production of enzymes liable to affect good heterologous expression, such as DNAses, proteases, or recombinases. Expression of foreign genes might be further enhanced by introducing genes encoding heterologous sigma factors (recognizing heterologous promoters) into the host genome (Gaida et al., 2015).
23– Transformed host cells should be easily detected. The sensitivity of bacteria to some specific antibiotic is generally used. If the cloning vector contains a resistance gene for this antibiotic (selection marker), only transformed cells are able to grow on a medium containing the antibiotic. Yeast transformants can be selected by functional complementation of an auxotrophic marker. For example, if a gene required for uracil production is disrupted in the host cell and if the cloning vector carries the functional gene, transformants can be recognized on the basis of their ability to grow on uracil-free medium.
24– A good host should also show no activity on the screening medium. Ideally, it should show as few enzymatic activities as possible for functional screening. Host cells can also be deprived by mutation of certain vital activities (e.g. DNA polymerase) to allow isolation of enzyme genes by functional complementation (Simon et al., 2009).
5.2. Prokaryotic hosts
25The most widely used bacterial host is the model bacterium Escherichia coli. This Gram- host is commonly used for library construction, because of its amenability to genetic engineering, its high transformation efficiency, and the availability of numerous genetic tools created for it. Several chemically competent or electro-competent E. coli strains are commercially available as well as efficient laboratory protocols to prepare competent E. coli cells. Although libraries are almost always constructed in E. coli, they can be screened in other bacteria if shuttle vectors are used. Examples of other bacterial species that have been used in (meta)genomic library screens include the proteobacterium Pseudomonas putida and its psychrophilic variant Pseudomonas antartica, the thermophile Thermus thermophilus, and the Gram+ bacteria Bacillus subtilis and Streptomyces lividans (reviewed in ; Taupp et al., 2011; Leis et al., 2013; Liebl et al., 2014). It can be assumed that close phylogenetic relationship between the expression host and the organism from which the foreign DNA derives should favor heterologous expression, and the efficiency of multi-host screenings in the identification of enzymes or molecules has indeed frequently been demonstrated. Despite the advantages of using different hosts, one should bear in mind that it always requires specific molecular tools (see host characteristics above).
5.3. Eukaryotic hosts
26Microbial eukaryotes can also be used as screening hosts. Yeasts such as Saccharomyces cerevisiae (whose genetics is well known and for which many genetic tools are available) and Pichia pastoris (with which excellent protein production yields are achieved) are widely used for their numerous advantages in high-level heterologous expression of genes encoding for enzymes (Liu et al., 2013). Such organisms combine the advantages of unicellular cells (easy to grow and manipulate genetically) with those of eukaryotic cells (better protein processing than in prokaryotes, allowing post-translational modifications and glycosylation) (Porro et al., 2005; Gündüz Ergün et al., 2015). An eukaryotic host should thus be the best choice for expressing genes from eukaryotic microbes. Yet as eukaryotic genomic DNA contains numerous introns, splicing of heterologous DNA could be problematic for the host. This explains why the use of cDNA libraries (obtained from RNA) is recommended for screening in eukaryotes (Kellner et al., 2011). The biggest limitations of using yeasts in functional screening could be poor recognition of heterologous promoters (especially if they are bacterial), low transformation yields (the libraries are then constructed in E. coli and screened in yeast), and the multiple enzymatic activities displayed by yeasts (the host should be mutated in all genes encoding enzymes of interest).
6. Functional screening of libraries
27Almost 7,000 enzyme types are currently listed in the BRENDA database (Chang et al., 2015). They are classified into six classes: oxidoreductases, transferases, hydrolases, lyases, isomerases, and ligases. As it is impossible to cover all the existing screening tests developed to date, next we show the most important steps in functional screening.
28Usually, isolated host-cell colonies containing plasmids with unique DNA inserts are recovered in 96-well plates (and stored at -80 °C in glycerol) for further screening. Otherwise, the colonies can be pooled in liquid culture, which is less fastidious at the outset but which can lead to generating biased libraries (some clones becoming dominant over or toxic towards others) and makes recovery of positive clones more laborious (as multiple copies of each clone will be present in the pool). How many clones one should screen depends on the size of the screened genome (easily estimated for gDNA but less obvious for eDNA and depending on the number of species present in the environmental sample), the DNA insert size range or the sizes and expression patterns of the genes sought (expression might depend on a vector promoter and/or RBS region and on the average distance between start codon and terminator) (for a review, see Gabor et al., 2004). According to Gabor et al. (2004), the number of clones screened should exceed 107, which is seldom the case (it is generally around 104-106), as generating and screening such huge libraries is probably too fastidious.
29Hydrolytic activities are generally assayed by growing the clones on agar plates or in well plates with liquid screening medium, and then detecting specific phenotypic traits. A color change occurring around the colony or in the well (directly or after addition of a second substrate), a clear halo, degradation of the medium, and fluorescence are the major visual observations used to detect an active clone. Such screens are easy to perform and do not require specific or high-technology material (unless colony-picking robots or microplate readers are used to speed up the screening). As the sensitivity of these phenotypic detection methods is usually low, they are used mostly when the aim is to scan the functional potential of a library (i.e. to scan for a broad range of enzymes), rather than to find a specific type of enzyme. One should bear in mind that a positive clone might appear negative because of inappropriate screening conditions, an inappropriate substrate, or because the enzyme is not been secreted (the phenotype may then appear later as a result of cell lysis).
30It is recommended to vary the screening conditions, to enhance the screening yield. Plates can easily be placed at different temperatures, for example, preferentially after overnight growth at the optimal growth temperature of the host. As enzymes might “prefer” some kind of substrates, prospection can be carried out on a broad range of natural, modified, and fully synthetic substrates (Leis et al., 2013). Well-known cofactors of the searched enzyme type can be added to the screening medium. Although varying the screening conditions may be time consuming, it can save time later by providing knowledge for the selection of clones with particular properties and/or for further characterization of the enzymes responsible for detected activities.
31To avoid the problem of non-secretion, one can mix cell lysates (obtained by enzymatic, physical, or chemical cell lysis) or permeabilized cells (obtained by treatment with a gentle detergent) with the screening substrate to enhance sensitivity (Taupp et al., 2011). Before cell lysis or permeabilization, the clones can be grown with the substrate of the enzyme sought, to enhance induction of a constitutional promoter of the gene of interest on the DNA insert. Likewise, UV- and heat-inducible vectors causing cell lysis have been developed to enhance extracellular activities (Xu et al., 2006; Li et al., 2007).
32As stated above, functional screening can also be done with a mutant host cell impaired in a vital enzymatic activity. The advantage of heterologous complementation is that the host cell is equipped to produce a protein having the same function, and that only clones producing the enzyme sought are viable (Ekkers et al., 2012). This method is very sensitive, but it is applied mostly to the identification of metabolites, as few vital enzymes are sought.
7. Conclusions
33The construction of libraries and their functional screening require experience and expertise. Choosing which environment to sample and which enzymes to seek is already a daunting task, given the immense diversity of both environments and enzymes. Once these decisions are made, success is likely to depend on other choices: the host cell used, the DNA insert size range, the targeted microorganisms, among others. In fact, there is no single perfect way to obtain high yields and to discover novel enzymes. In most cases, functional (meta)genomics with adequate adjustments should lead to the identification of novel enzymes. The best way to obtain a wide diversity of novel enzymes is probably to combine different strategies, such as working with cultivable and non-cultivable organisms, using sequence- and function-based approaches, performing multi-host screenings, and constructing libraries in both plasmids and fosmids.
Bibliographie
Aakvik T. et al., 2009. A plasmid RK2-based broad-host-range cloning vector useful for transfer of metagenomic libraries to a variety of bacterial species. FEMS Microbiol. Lett., 296, 149-158.
Adrio J.L. & Demain A.L., 2014. Microbial enzymes: tools for biotechnological processes. Biomolecules, 4, 117-139.
Berasategui A., Shukla S., Salem H. & Kaltenpoth M., 2015. Potential applications of insect symbionts in biotechnology. Appl. Microbiol. Biotechnol., 100, 1567-1577.
Bleidorn C., 2015. Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers., 2000, 1-8.
Borglin S. et al., 2012. Application of phenotypic microarrays to environmental microbiology. Curr. Opin. Biotechnol., 23, 41-48.
Borkar S., 2015. Alkaliphilic bacteria: diversity, physiology and industrial applications. In: Borka S., ed. Bioprospects of coastal eubacteria. Springer International Publishing, 59-83.
Burke C., Kjelleberg S. & Thomas T., 2009. Selective extraction of bacterial DNA from the surfaces of macroalgae. Appl. Environ. Microbiol., 75, 252-256.
Chang A. et al., 2015. BRENDA in 2015: exciting developments in its 25th year of existence. Nucleic Acids Res., 43, D439-446.
Cobb R.E., Chao R. & Zhao H., 2013. Directed evolution : past, present and future. AIChE J., 59, 1432-1440.
de Lourdes Moreno M., Pérez D., García M.T. & Mellado E., 2013. Halophilic bacteria as a source of novel hydrolytic enzymes. Life, 3, 38-51.
De Monte S., Soccodato A., Alvain S. & d’Ovidio F., 2013. Can we detect oceanic biodiversity hotspots from space? ISME J., 7, 2054-2056.
Denard C.A., Ren H. & Zhao H., 2015. Improving and repurposing biocatalysts via directed evolution. Curr. Opin. Chem. Biol., 25, 55-64.
Dorofeev A.G. et al., 2014. Approaches to cultivation of “nonculturable” bacteria: Cyclic cultures. Microbiology, 83, 450-461.
Ekkers D.M., Cretoiu M.S., Kielak A.M. & Elsas J.D., 2012. The great screen anomaly – a new frontier in product discovery through functional metagenomics. Appl. Microbiol. Biotechnol., 93, 1005-1020.
Faure D. & Joly D., 2015. Next-generation sequencing as a powerful motor for advances in the biological and environmental sciences. Genetica, 143, 129-132.
Ferrer M. et al., 2016. Estimating the success of enzyme bioprospecting through metagenomics: current status and future trends. Microb. Biotechnol., 9, 22-34.
Gabor E.M., Alkema W.B.L. & Janssen D.B., 2004. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ. Microbiol., 6, 879-886.
Gaida S.M. et al., 2015. Expression of heterologous sigma factors enables functional screening of metagenomic and heterologous genomic libraries. Nat. Commun., 6, 7045.
Gavrilescu M. & Chisti Y., 2005. Biotechnology – a sustainable alternative for chemical industry. Biotechnol. Adv., 23, 471-479.
Gündüz Ergün B. & Çalık P., 2015. Lignocellulose degrading extremozymes produced by Pichia pastoris: current status and future prospects. Bioprocess Biosyst. Eng., 39(1), 1-36.
Handelsman J., 2004. Metagenomics : Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev., 68, 669-685.
He Z., Zhu Y. & Gu H., 2013. A new method for the determination of critical polyethylene glycol concentration for selective precipitation of DNA fragments. Appl. Microbiol. Biotechnol., 97, 9175-9183.
Highlander S., 2014. Culturing. In: Nelson K.E., ed. Encyclopedia of metagenomics. New York, USA: Springer, 1-4.
Jung D. et al., 2014. Application of a new cultivation technology, I-tip, for studying microbial diversity in freshwater sponges of Lake Baikal, Russia. FEMS Microbiol. Ecol., 90, 417-423.
Kakirde K.S., Parsley L.C. & Liles M.R., 2010. Size does matter: Application-driven approaches for soil metagenomics. Soil Biol. Biochem., 42, 1911-1923.
Kamagata Y., 2015. Keys to cultivating uncultured microbes: elaborate enrichment strategies and resuscitation of dormant cells. Microbes Environ., 30, 289-290.
Kato C., 2012. Microbiology of piezophiles in deep-sea environments. In: Anitori R.P., ed. Extremophiles: microbiology and biotechnology. Norfolk, UK: Caister Academic Press, 233.
Kellner H., Luis P., Portetelle D. & Vandenbol M., 2011. Screening of a soil metatranscriptomic library by functional complementation of Saccharomyces cerevisiae mutants. Microbiol. Res., 166, 360-368.
Kuzyakov Y. & Blagodatskaya E., 2015. Microbial hotspots and hot moments in soil: Concept & review. Soil Biol. Biochem., 83, 184-199.
Leis B., Angelov A. & Liebl W., 2013. Screening and expression of genes from metagenomes. Adv. Appl. Microbiol., 83, 1-68.
Li S. et al., 2007. A set of UV-inducible autolytic vectors for high throughput screening. J. Biotechnol., 127, 647-652.
Liebl W. et al., 2014. Alternative hosts for functional (meta)genome analysis. Appl. Microbiol. Biotechnol., 98, 8099-8109.
Liu L. et al., 2013. How to achieve high-level expression of microbial enzymes: Strategies and perspectives. Bioengineered, 4, 212-223.
Martin M., Portetelle D., Michel G. & Vandenbol M., 2014. Microorganisms living on macroalgae: diversity, interactions, and biotechnological applications. Appl. Microbiol. Biotechnol., 98, 2917-2935.
Martinez A. et al., 2004. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl. Environ. Microbiol., 70, 2452-2463.
Morgavi D.P., Kelly W.J., Janssen P.H. & Attwood G.T., 2013. Rumen microbial (meta)genomics and its application to ruminant production. Animal, 7, 184-201.
Narihiro T. et al., 2014. The combination of functional metagenomics and an oil-fed enrichment strategy revealed the phylogenetic diversity of lipolytic bacteria overlooked by the cultivation-based method. Microbes Environ., 29, 154-161.
Packer M.S. & Liu D.R., 2015. Methods for the directed evolution of proteins. Nat. Rev. Genet., 16, 379-394.
Pham V.H.T. & Kim J., 2012. Cultivation of unculturable soil bacteria. Trends Biotechnol., 30, 475-484.
Porro D., Sauer M., Branduardi P. & Mattanovich D., 2005. Recombinant protein production in yeasts. Mol. Biotechnol., 31, 245-259.
Prosser G.A., Larrouy-Maumus G. & de Carvalho L.P.S., 2014. Metabolomic strategies for the identification of new enzyme functions and metabolic pathways. EMBO Rep., 15, 657-669.
Raddadi N. et al., 2015. Biotechnological applications of extremophiles, extremozymes and extremolytes. Appl. Microbiol. Biotechnol., 99, 7907-7913.
Raval V.H., Purohit M.K. & Singh S.P., 2013. Diversity, population dynamics and biocatalytic potential of cultivable and non-cultivable bacterial communities of the saline ecosystems. In: Trincone A., ed. Marine enzymes for biocatalysis. Sources, biocatalytic characteristics and bioprocesses of marine enzymes. Cambridge, UK: Woodhead Publishing Limited, 165-189.
Rhoads A. & Au K.F., 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinf., 13, 278-289.
Riquelme C., Rojas A., Florest V. & Correatj J.A., 1997. Epiphytic bacteria in a copper-enriched environment in Northern Chile. Mar. Pollut. Bull., 34, 816-820.
Roberts S.M., Turner N.J., Willetts A.J. & Turner M.K., 1995. Introduction to biocatalysis using enzymes and microorganisms. Cambridge, UK: Cambridge University Press, 34-79.
Rodriguez-Valera F., 2014. Fosmid system. In: Nelson K.E., ed. Encyclopedia of metagenomics. New York, USA: Springer, 1-5.
Schofield M.M. & Sherman D.H., 2013. Meta-omic characterization of prokaryotic gene clusters for natural product biosynthesis. Curr. Opin. Biotechnol., 24, 1151-1158.
Shizuya H. et al., 1992. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. U.S.A., 89, 8794-8797.
Simon C., Herath J., Rockstroh S. & Daniel R., 2009. Rapid identification of genes encoding DNA polymerases by function-based screening of metagenomic libraries derived from glacial ice. Appl. Environ. Microbiol., 75, 2964-2968.
Steinert G. et al., 2014. Application of diffusion growth chambers for the cultivation of marine sponge-associated bacteria. Mar. Biotechnol., 16, 594-603.
Taupp M., Mewis K. & Hallam S.J., 2011. The art and design of functional metagenomic screens. Curr. Opin. Biotechnol., 22, 465-472.
Terrón-González L., Medina C., Limón-Mortés M.C. & Santero E., 2013. Heterologous viral expression systems in fosmid vectors increase the functional analysis potential of metagenomic libraries. Sci. Rep., 3, 1107.
Thomas T., Gilbert J. & Meyer F., 2012. Metagenomics - a guide from sampling to data analysis. Microb. Inf. Exp., 2, 3.
Ufarte L., Potocki-Veronese G. & Laville E., 2015. Discovery of new protein families and functions: new challenges in functional metagenomics for biotechnologies and microbial ecology. Front. Microbiol., 6, 1-10.
Vester J.K., Glaring M.A. & Stougaard P., 2015. Improved cultivation and metagenomics as new tools for bioprospecting in cold environments. Extremophiles, 19, 17-29.
Walker A.W., Duncan S.H., Louis P. & Flint H.J., 2014. Phylogeny, culturing, and metagenomics of the human gut microbiota. Trends Microbiol., 22, 267-274.
Wang L., Nasrin S., Liles M. & Yu Z., 2014. Use of bacterial artificial chromosomes in metagenomics studies, overview. In: Nelson K.E., ed. Encyclopedia of metagenomics. New York, USA: Springer.
Warnecke F. & Hess M., 2009. A perspective: metatranscriptomics as a tool for the discovery of novel biocatalysts. J. Biotechnol., 142, 91-95.
Xu L. et al., 2006. Heat-inducible autolytic vector for high-throughput screening. BioTechniques, 41, 319-323.
Yarza P. et al., 2014. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol., 12, 635-645.
Zhou J., Bruns M.A. & Tiedje J.M., 1996. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol., 62, 316-322.
